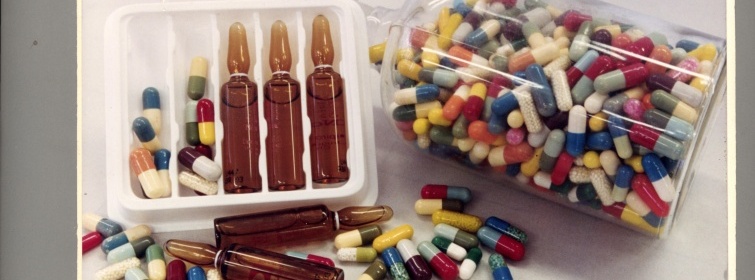
El reconocido estudio Ferrere nos explica los alcances del nuevo reglamento que regula el registro, producción, exportación, importación y comercialización de medicamentos través del Decreto 18/020 aprobado por el Poder Ejecutivo.
El informe lleva la firma de los profesionales Bruno Bertolotti, Leticia Aguiar, y Agustín Marchesano.
El Decreto actualiza la normativa de hace 20 años dejando sin efecto -prácticamente en su totalidad- el reglamento del año 1999 (Decreto Nº 324/999).
A continuación, incluimos un resumen de los principales aspectos y novedades de la nueva reglamentación, que será de aplicación a la totalidad de los medicamentos, sin perjuicio del cumplimiento de requisitos adicionales establecidos por otras normas especiales para cada clase de medicamento en particular.
Las empresas instaladas en Zona Franca o Puerto Libre deberán habilitarse: se establece expresamente que las empresas ubicadas en dichos espacios deberán estar habilitadas ante el Ministerio de Salud Pública (MSP) si solicitan el registro de un medicamento.
Nuevas categorías de registro: se establecen, además del registro sanitario en el MSP para importación y/o comercialización en plaza, nuevas categorías de registro dependiendo de la actividad a realizar:
Registro para exportación: se crea un procedimiento abreviado para el registro de productos elaborados en Uruguay con fines exclusivos de exportación.
Registro para condiciones especiales: refiere a la posibilidad de que el MSP registre medicamentos de oficio a efectos de importarlos directamente, cuando resulta necesario para el cumplimiento de planes o programas de salud.
Se elimina la aprobación ficta del registro de ciertos medicamentos: a diferencia de lo establecido por el decreto anterior, no se prevé ningún plazo máximo para la evaluación del trámite de registro y su aprobación ficta.
Nuevos plazos para solicitar información adicional en el trámite de registro: en caso que el MSP solicite aclaraciones o información adicional, el plazo para responder será de 30 días corridos desde la notificación. Los pedidos podrán repetirse hasta tres veces y en cada caso el MSP contará con 120 días corridos (salvo en caso de medicamentos intercambiables o necesidad de consulta externa) para la evaluación de las respuestas.
No se prevé trámite urgente: salvo en casos excepcionales, las solicitudes de registro serán evaluadas por orden de ingreso. El orden podrá alterarse en caso en que el producto sea necesario para cumplir con planes o programas de salud.
Certificados GMP y CPP: se establece que, para el registro, los importadores deberán presentar certificado de Buenas Prácticas de Manufactura (GMP) del fabricante/s de origen y Certificado de Producto Farmacéutico (CPP). Asimismo, en caso de no contar con el GMP, el MSP podrá realizar inspecciones de verificación del cumplimiento de GMP a costo del interesado.
Presentación de documentación en español y completa: salvo excepciones, toda documentación deberá ser presentada en idioma español y en forma completa, no admitiéndose el agregado de documentación faltante. En caso de falta de documentación o que la misma no sea acorde a la normativa vigente, se denegará la solicitud de registro y se deberá esperar 180 días para realizar una nueva solicitud.
Información confidencial: se establece expresamente que la información técnica que se presente en el trámite de registro será considerada confidencial.
Lanzamiento (comercialización): si bien no existen modificaciones significativas, se agrega la exigencia de comunicar el lanzamiento con 10 días hábiles de anticipación al mismo.
Interrupción de abastecimiento de mercado: se prevé que la comercialización de un medicamento solo podrá interrumpirse en carácter excepcional, por un plazo no superior a 30 días, debiendo comunicarse tal situación al MSP. En caso que se prevea que la interrupción será superior a 30 días y se trate de medicamentos que requieren control profesional, adicionalmente, deberá declararse como se asegurará la disponibilidad de los productos para pacientes en tratamiento.
Renovación de registro: será obligatoria la presentación de la solicitud de renovación de registro con una antelación máxima de 90 días a la fecha de vencimiento del registro vigente.
Caducidad y baja del registro: si se detecta la imposibilidad de comercializar el medicamento por tres años consecutivos, el registro caducará en forma automática.
A diferencia de lo que establecía la reglamentación anterior, la solicitud de baja del registro deberá fundamentarse y ser autorizada por el MSP.
Suspensión o cancelación del registro: el nuevo reglamento modifica las causales por las cuales se podrá suspender o cancelar el registro del medicamento, incluyendo: cuando se detecten problemas vinculados a su uso o un apartamiento de calidad o a partir de alertas emitidas por autoridades reguladoras extranjeras u organismos internacionales.
Ingreso y comercialización de productos no registrados: se prevén dos situaciones en las cuales se autorizará el ingreso y comercialización de medicamentos no registrados en el país:
Cuando exista un medicamento con: (a) un fin medicinal urgente y (b) no exista en el país una alternativa terapéutica. En este caso, será necesario: i) solicitud firmada del médico tratante y por la Dirección Técnica de la institución a la que pertenece el paciente; ii) consentimiento informado del paciente.
Cuando sean destinados a investigación científica o ensayos clínicos.
Se modifican las farmacopeas de referencia: serán consideradas de referencia la “Farmacopea Europea” y la “United States Pharmacopeia” de los últimos tres años. Se eliminan las Farmacopeas de Argentina y Brasil y el Codex Francés, previstas por el reglamento anterior.
Donaciones: anteriormente se preveía la donación únicamente a entidades benéficas. El nuevo reglamento no limita el beneficiario. La donación deberá ser autorizada por el MSP y previo a su distribución el medicamento será analizado por la Comisión de Control de Calidad de Medicamentos.
Rotulado y Prospecto: el MSP definirá mediante ordenanza los requisitos generales.
Medicamentos Controlados: la leyenda “Medicamento Controlado” solo deberá incluirse en el envase secundario, ya no en el envase primario, prospecto y etiqueta.
VIGENCIA
El Decreto establece que entrará en vigencia a los 90 días de su publicación en el Diario Oficial y se aplicará a los nuevos trámites que ingresen. Sin perjuicio de ello, será de aplicación inmediata a los trámites de registro ya ingresados en la Autoridad Reguladora Nacional de Medicamentos la exigencia de presentar GMP y CPP. A la fecha de publicación de este artículo, el decreto aún no había sido publicado.
Firman: Bruno Bertolotti, Leticia Aguiar, y Agustín Marchesano.
Fuente Imagen: cccm.org.uy






{kind=link}